Serving 200+ Researchers
on omics research
600+ Software/Tools
extensive software library
387,000+ Jobs Completed
over the past 12 months
High Performance Computing Cluster (HPCF3)
🖥️ HPCF3 GPU Trial Promotion – Now open for FREE!
➡️ Click here to know more details.
Hardware and Access
Hardware and Access
🖥️HPCF3 Cluster Overview
The HPCF3 login node is accessible via SSH using the hostname:
hpcf3.cpos.hku.hk
Click here for more details.
🧰Operating System & Software
All HPCF3 servers run on Rocky Linux 8.
A broad range of commonly used bioinformatics software is preinstalled and available to all users.
⚙️Services
Users submit jobs through OpenPBS, the cluster’s job scheduling system.
Execution is carried out on compute nodes via PBS jobs only.
➡️ Direct login to compute nodes is not permitted.
👤User Account Setup
To request a user account, contact itsupport.cpos@hku.hk.
- All account applications are subject to CPOS approval.
- Approved users receive a Linux account to access the cluster.
- Each account comes with 100 GB default storage; additional quota can be requested (subject to availability and charges).
- Each account is assigned to a named individual — account sharing is strictly prohibited.
- While storage systems are protected by hardware redundancy, users are strongly advised to maintain their own backups regularly.
- Users should be familiar with the Linux/UNIX environment. A reference guide is available from ITS.
🔐Cluster Login
Users may connect remotely via SSH using clients such as PuTTY, available at: http://www.chiark.greenend.org.uk/~sgtatham/putty/
Connection Details
To connect to the login node of HPCF3, please use an SSH client to open a SSH terminal session with the following:
Hostname: hpcf3.cpos.hku.hk Username:
🔒Network Requirement
For security reasons, HPCF3 servers are accessible only from within the HKU network.
-
Off-campus users must first connect through HKUVPN.
Details: https://its.hku.hk/services/network-connectivity/hkuvpn/
📦Data Transfer
🪟 Windows Users
For Windows users there are several free, GUI-based secure file transfer clients, such as WinSCP or FileZilla. You could use these clients with simply drag and drop files between the cluster front-end node and your desktop or laptop.
🍏 Mac / 🐧 Linux Users
For a Mac or Linux, the command-line scp client is typically installed with the operating system. You can access it through a terminal running on your local machine. For example, suppose you have a Mac, and a local file called myfile on your mac that you want to copy to your home directory on the cluster.
Example:
This copies myfile into the directory mydir/ under your HPCF3 home directory.
As with any Unix command that takes files or directories as arguments, the file or directory for either the source or destination can be specified as either an absolute path (one that begins with a slash) or a relative path (one that does not). For local files or directories, relative paths are relative to the current working directory. For remote files or directories, relative paths are relative to a home directory on the remote host.
📡Bulk Data Transfer Guidelines
To avoid impacting campus network performance:
- Transfers larger than 30 GB to/from outside the HKU network should be done outside office hours.
- For transfers exceeding 500 GB, please notify itsupport.cpos@hku.hk in advance so we can coordinate with ITS.
⚠️ Failure to provide notice for large transfers may result in ITS temporarily blocking cluster servers from the Internet.
Charges
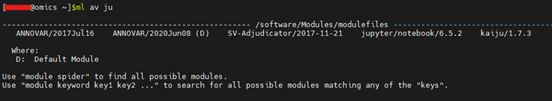
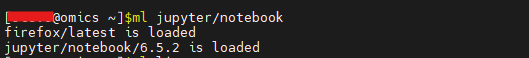
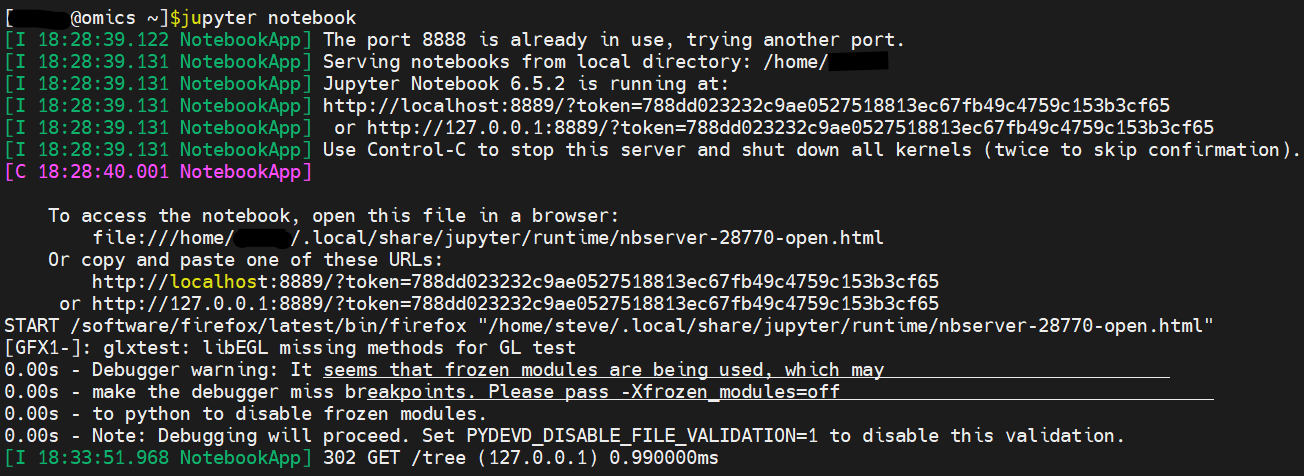
Environment Modules
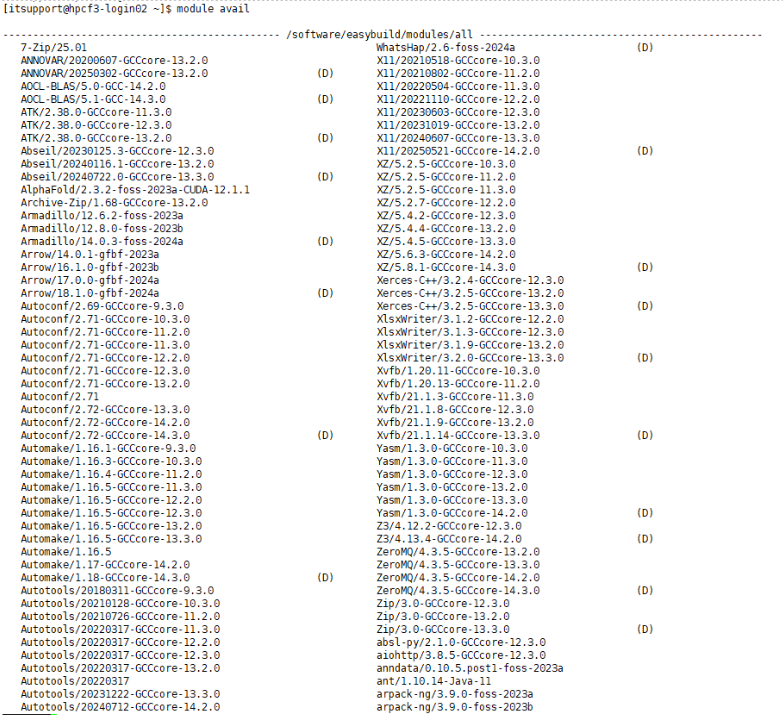
Installed Software
Job Scheduling
High Performance Computing Cluster (HPCF2)
Hardware and Access
Hardware and Access
HPCF2 cluster
The master node is Omics which can be accessed by SSH with hostname hpcf2.cpos.hku.hk.
The HPCF2 cluster consists of 8 compute nodes with system configuration listed below:
| Server | CPU Brand & Model | No of CPU | Cores per CPU | Physical Cores per Server | CPU Threads per Server | RAM (GB) |
| hpch01 | Intel Xeon E5-2650 v4 2.2GHz | 2 | 12 | 24 | 48 | 256 |
| hpch02 | Intel Xeon E5-2650 v4 2.2GHz | 2 | 12 | 24 | 48 | 256 |
| hpch03 | Intel Xeon E5-2650 v4 2.2GHz | 2 | 12 | 24 | 48 | 256 |
| hpch04 | Intel Xeon E5-2650 v4 2.2GHz | 2 | 12 | 24 | 48 | 256 |
| hpch05 | Intel Xeon E5-2650 v4 2.2GHz | 2 | 12 | 24 | 48 | 256 |
| hpch06 | Intel Xeon E5-2650 v4 2.2GHz | 2 | 12 | 24 | 48 | 256 |
| hpch07 | Intel Xeon E5-2650 v4 2.2GHz | 2 | 12 | 24 | 48 | 256 |
| hpch08 | Intel Xeon E5-2683 v4 2.1GHz | 2 | 16 | 32 | 64 | 512 |
| Total: | 200 | 400 | 2,304 |
Operating System and System Software
All servers in HPCF2 are running CentOS 7 Linux Operating System and major bioinformatics software have been installed and ready for use by all users.
Cluster Nodes
Master Nodes
Services
- Compile/Run command-line programs in interactive mode via SSH
- Submit batch jobs to the job queue system
- File transfer via sftp client like Filezilla
Control Measures
The master node shall be used for the services above while users’ analysis jobs shall be executed at the compute nodes by job submission to the job queue system. Particularly, the master node shall not be used to execute those resource hungry jobs. Therefore, the following control measures are implemented at the master nodes:
- The maximum total CPU usage of all the user’s jobs at any one time is 400% (i.e. running four jobs concurrently that each thread consumes 100% CPU time or a single job with 4 threads each consume 100% CPU time).
- The maximum running time of each user’s job is 10 minutes.
- The maximum total amount of main memory usage of all the user’s jobs at any one time is 10GB.
The master node will check if the limits are exceeded for each user and will terminate the user jobs if the limits are exceeded in descending order of usage until the resource usage is brought down to the accepted limits. Notification emails will be automatically sent to the user with the details of the processes that are terminated.
Compute Nodes
Services
- User can submit PBS jobs for execution on the compute nodes from the master node through the job scheduling system. Therefore, user cannot login to the compute node directly for job execution.
User Account Setup
- Please contact itsupport.cpos@hku.hk for user account setup.
- Account approval is subject to consideration by CPOS.
- Upon approval of account application, each user is allocated a Linux user account for access to the cluster.
- Each user is allocated a Linux user account with 100GB disk space by default initially. User may apply for extra disk space subject to availability and charges.
- Each user account should be used for and held responsible to a named person. NO account sharing is allowed.
- User may apply for extra disk quota subject to availability and charges.
- Although the storage is protected by hardware redundancy, user is highly recommended to do his/her own regular backup of important files from the cluster to a local disk for peace of mind.
- The user shall be familiar with Linux/UNIX software environment. You may refer to the Unix user guide prepared by ITS at here as reference.
Cluster Login
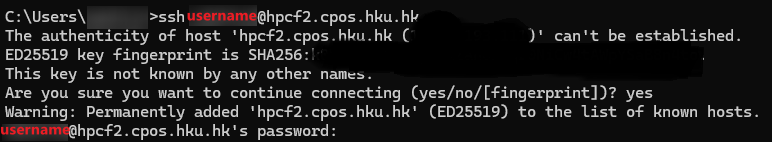
User may remotely connect to a command line terminal of HPCF2 via SSH using SSH clients like PUTTY (Link to SSH client: http://www.chiark.greenend.org.uk/~sgtatham/putty/).
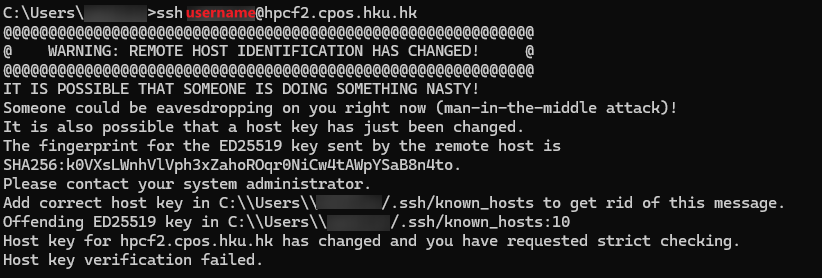
To connect to the master node of HPCF2, please use an SSH client to open a SSH terminal session with the following:
hostname: hpcf2.cpos.hku.hk
Login with the same user account as in the current HPCF.
N.B. For security reasons, the servers can only be connected from machines in the HKU network. For machines outside of HKU network, user has to connect to HKUVPN before connecting to the servers. Please see the details at https://its.hku.hk/services/network-connectivity/hkuvpn/
Data Transfer
For Windows users there are several free, GUI-based secure file transfer clients, such as WinSCP or FileZilla (http://filezilla-project.org/). You could use these clients with simply drag and drop files between the cluster front-end node and the your desktop or laptop.
For a Mac or Linux, the command-line scp client is typically installed with the operating system. You can access it through a terminal running on your local machine. For example, suppose you have a Mac, and a local file called myfile on your mac that you want to copy to your home directory on the cluster.
On a Mac/Linux PC, start the Terminal application and type
scp myfile userid@hpcf2.cpos.hku.hk:mydir/
That would copy the local file myfile to the subdirectory mydir/ under your home directory on the cluster and keeps the filename myfile. As with any Unix command that takes files or directories as arguments, the file or directory for either the source or destination can be specified as either an absolute path (one that begins with a slash) or a relative path (one that does not). For local files or directories, relative paths are relative to the current working directory. For remote files or directories, relative paths are relative to a home directory on the remote host.
Bulk Data Transfer
In order not to affect other users in the HKU campus network, it is suggested by the HKU Information Technology Services that network transfer of bulk data (>30GB) between the cluster and machines outside the HKU Network should be scheduled in non-office hours. If the data to be transferred is of more than 500GB, prior notice should be sent to itsupport.cpos@hku.hk such that we can relay it to ITS for the planned network traffic. Failure to do so may result in ITS blocking our servers from reaching the Internet.
Decommission
HPCF2 will be fully decommissioned by Oct-2026
Charges
Environment Modules
Installed Software
Job Scheduling
FAQ

Contact


