Project Planning
Data analysis is crucial for any NGS project and we recommend advance planning. For researchers needing bioinformatics support from us, we strongly encourage you discussing of your projects with us at an early stage of project planning so as to evaluate feasibility and set proper expectations.
What to expect
Our FREE consultation will help you decide on the choices of sequencing technology (see table below), length of read, throughput required (amount of coverage) and analysis methodologies etc. All of these will play an important role in answering your specific scientific questions.
Below table lists some of the typical projects with suitable technologies, but we may discuss any other.
Contact us NOW to discuss your project!
| Application | Illumina Sequencing Technologies, sequencing by synthesis | Single molecule, real-time sequencing technology (SMRT) | Possible Questions Being Asked | |
| NovaSeq | MiSeq | PacBio | ||
| Exome sequencing | +++ | + | - | Are there novel mutations in the exonic sequences? |
| RNA-Seq | +++ | ++ | + | What genes are differentially expressed in my samples? Are there different splicing patterns between my samples? |
| ChIP-Seq | +++ | ++ | - | What sequences do my protein binds to? |
| de novo whole genome sequencing | +++ | +++ | +++ | How does the genome of this organism look like? |
| Resequencing | +++ | +++ | + | What are the sequence differences between my isolate of bacteria/virus/plasmid compare to the common strains? |
| Metagenomics | +++ | ++ | + | What micro-organisms are present in my environmental sample? What are the different virus strains in my culture? |
| MicroRNA profiling | +++ | ++ | - | What microRNAs are expressed, or differentially expressed in my samples? |
| Methylation analysis | +++ | ++ | + | Which sites are methylated in the genome? Is the methylation pattern different between my samples? |
Workflow
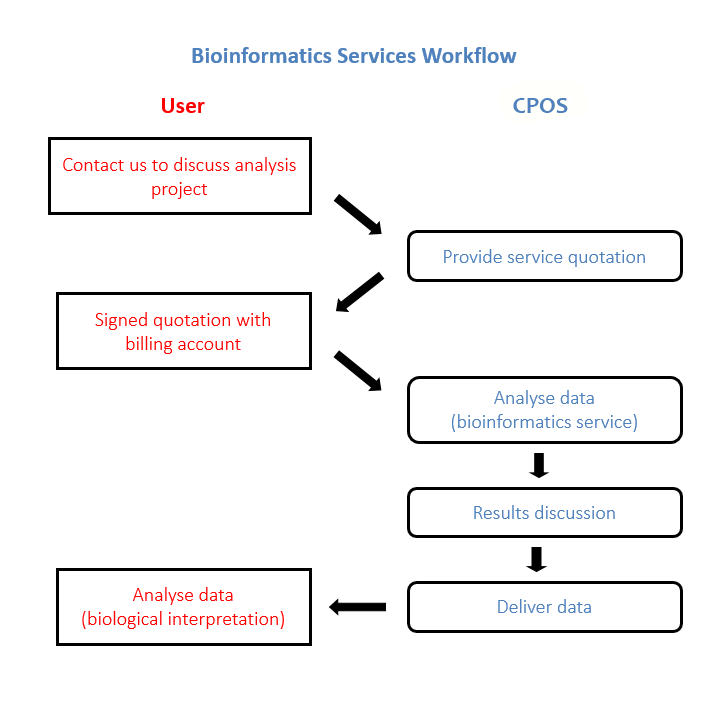
Service Charges
Most bioinformatics analyses are unique so service charge vary depending on the time and resources required to manage your particular project.
For budgeting purpose, after we learn of your needs, we will provide a quotation of estimated cost to you promptly.
As an academic, non-profit-making core facility, we offer a highly competitive price as compared to many other service providers.
*IMPORTANT:* To qualify for HKU pricing, the investigator must be a regular employee of HKU AND payment must be made from an internal HKU financial account that qualifies for internal transfer. Overhead charge is required for all incoming external funding.
Service Options
At CPOS, the bioinformatics team works closely with the NGS laboratory team to provide various service options to suit your needs.
With the exception of sequencing run service, sequencing data are QC prior to being released to users.
For more details about the specific deliverables according to applications, please review Data Deliverables section.
| Service Type | Library Prepared at CPOS? | Sequencing Handled by CPOS? | Bioinformatics Deliverables | ||||
| Raw Sequencing Data (de-multiplexed) | Sequencing Report | FastQC Report | Analysis Report and other Deliverables | Intermediate Analysis Files | |||
| Sequencing run service | No | Yes | √ | ||||
| Full service | Yes | Yes | √ | √ | √ | ||
| Full service with standard analysis | Yes | Yes | √ | √ | √ | √ | √ |
| Data analysis only | No | No | √ | √ |
Data Collection
Overview
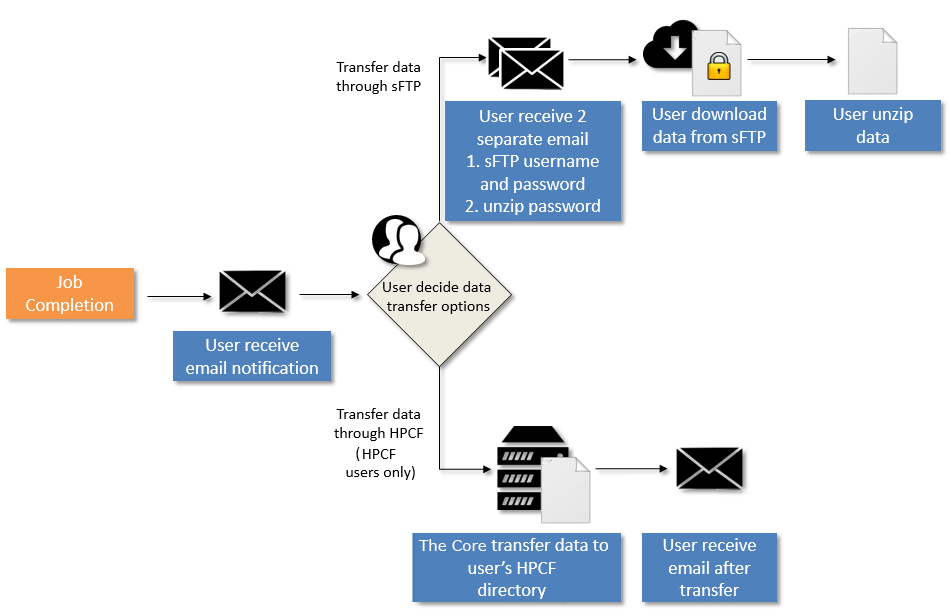
Once data is available, users will be notified via email. Two data transfer options are available:
(I) For all users, data could be downloaded via sFTP server.
- User receives sFTP username and password through email, unzip password will be provided in a separate email.
- User downloads data from sFTP server. Instructions can be found here.
- To unzip files, user can use 7zip or winrar .
(II) For HPCF* users only, data could be transferred directly to user-specified HPCF directories.
- User provides HPCF folder address to the Core.
- The Core notifies user upon completing data transfer through email.
In compliance with the centre’s data protection scheme, analysed data are compressed and encrypted prior to delivery via sFTP server. Username and password to access sFTP server and password to unzip files are provided in separate email for added security. Data could be unzipped using 7zip or winrar.
Due to limited server hard disk space, data will only be kept for 1 month after delivery. Data will then be removed from our servers without prior notice.
Please ensure that you keep a copy of the data (analysis results and all intermediate files) securely and clearly identified for future reference.
*For more information about HPCF, please visit the HPCF section.
Data Collection Workflow
How to Download Files from sFTP Server
Summary
This section shows how to transfer files from sFTP Server by setting up and logging into Filezilla client as the preferred FTP client. The downloaded files will be split into multiplex files if the size is larger than 250 GB. Please remember to download the md5sum file in the same folder to verify the integrity of the downloaded file later. Due to the instability of Wi-Fi, Wi-Fi is not recommended to use for the download.
Procedures
- Download and install Filezilla client (https://filezilla-project.org). Note: please download the Filezilla client and NOT the file Filezilla Server.
- Open Filezilla. Enter the following information into the Quickconnect bar located at the top of the window.
- Host: Your given host
- Username: Your given username
- Password: Your given password
- Port number: 22
(The above information can be obtained from the email sent by the bioinformatics service team)

- Click on Quickconnect or press Enter to connect to the server.

- On first login, click OK to accept the security certificate about an unknown host key.

- Click on the file that you wish to download from sFTP Server (window on the right) and then move the file to the destination location on your computer (window on the left). Please note that you will need to click and hold the mouse button during this drag and drop action.
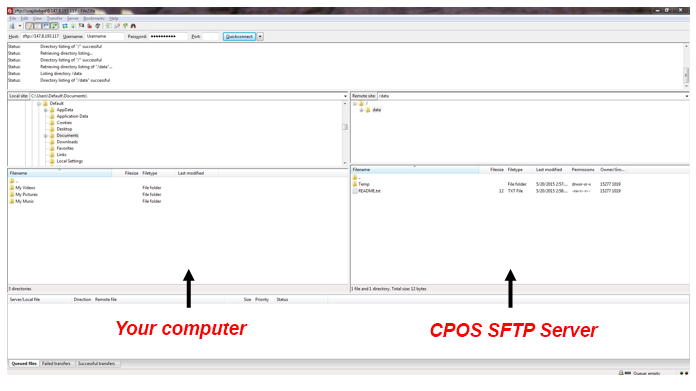
- Upon “dropping” the file into your computer, you will see that the file transfer is in progress (see screen shot below). Please wait for the file transfer to be completed and this will take a while for files that is large in size. Note that the number in the brackets denotes how many files are to be transferred.

- Once the file transfer completed successfully, you will see numbers in the “Successful transfers” tab (see screen shot below). Note that the number in the brackets denotes how many files are transferred successfully.

- You can close Filezilla client when all files are downloaded successfully and proceed to verify the downloaded file using md5sum (Refer to next section). Please ensure that you follow the next step carefully to confirm that the download is successful.
How to Check md5sum of Downloaded Files
Summary
This section shows how to verify the integrity of the downloaded file using the MD5 (Message-Digest algorithm 5) hash value by WinMD5Free.
Procedures
- Download and unzip WinMD5Free (http://www.winmd5.com).
- Open the WinMD5.exe in the unzipped folder.
- Click on Browse and choose the zip file you have downloaded from sFTP Server, the MD5 checksum value will be computed and shown in Current file MD5 checksum value. Please wait patiently as this process will take a while (up to an hour or more) for file that is large in size.

- Open the md5sum file downloaded from sFTP server in Notepad. Copy the MD5 checksum value and paste into Original file MD5 checksum value in WinMD5Free, then click Verify.
Notepad:  WinMD5Free:
WinMD5Free:
- A window will pop up and show “Matched!” if the download from sFTP server is success. If “NOT Matched!” is shown, please download the file again from our sFTP server.

- After confirming that the file is downloaded successfully, you can proceed to unzip the file using 7-zip if needed (Refer to next section).
How to Unzip Password Protected Files Downloaded from sFTP Server
Summary
This section shows how to unzip the password protected files downloaded from our sFTP server using 7-Zip. Please remember to download all of the split zip files BEFORE attempting to unzip the files.
WARNING: Please ensure you have sufficient disk space to hold all the un-compress data.
Procedures
- Download and install 7zip (http://www.7-zip.org).
- Right click the zip.001 file you have downloaded from SFTP server, then go 7-Zip and click Extract Here.

- A window will pop up and ask for the password. Please enter the password that we have provided through email and click OK.

- An unzipped folder will be extracted at the same location as the original file. Please note that only zip.001 need to be extracted and the remaining zip files (if any) will be unzipped into a single folder.
Data Deliverables
Typical Deliverables
Depending on the sequencing technology, project type and amount of bioinformatics support required, the deliverables will differ. In general this includes:
- Sequencing Report (MS Word file) – a written report documenting the performance of the sequencing job.
- FastQC Report (HTML file) – reports generated by a NGS data quality control tool which can be viewed on a web browser (Illumina Sequencing only). Visit the author’s website for details.
- Analysis Report and Results (various file types) – reports summarizing the analyzed data and the analysis pipeline applied. Project-specific analysis result files are also included (see below table).
- Intermediate analysis files (various file types) – files that are created throughout analysis, e.g. filtered high-quality reads in Fastq format. Deliverables depend on project type (see below table).
Please choose the type of sequencing technology below to view the deliverables.
WP Table Builder


Illumina NovaSeq / MiSeq Sequencing – Specific Deliverables Based On Project Type
For analysis needs beyond our routine analysis pipeline (standard deliverables), customized assistance could be provided (custom deliverables). Please note that the list below is by no means an exhaustive list of deliverables possible. Do contact us if you cannot locate what you need. Final deliverables are subjected to mutual agreement between CPOS and users.
| Type of Project | Standard Deliverables | Custom Deliverables |
| RNA-Seq (mRNA) | - Alignment Files in BAM format - Gene / Transcript Expression Level File in MS Excel format - *List of Differentially Expressed Gene / Transcript in MS Excel format (includes integration into Partek Flow for downstream pathway analysis) - PCA plot in HTML format (at least 2 samples) *Includes 1 pairwise comparison for every sample submitted for sequencing. Additional comparisons are welcome, please contact us for details. | - List of Annotated SNP / INDEL in MS Excel format - Alternative Splicing Patterns in MS Excel format - Fusion Gene File in MS Excel format - Novel Exon File in MS Excel format - Others (open for discussion) |
| RNA-Seq (miRNA) | - Alignment Files in BAM format (known miRNA) - Alignment Files in BAM format (to other known RNAs, i.e. snRNA, snoRNA and etc.) - miRNA Expression Level File in MS Excel format - *List of Differentially Expressed miRNA in MS Excel format *Includes 1 pairwise comparison for every sample submitted for sequencing. Additional comparisons are welcome, please contact us for details. | - Target Prediction of miRNA - Novel miRNA File in MS Excel format - Expression level of other known RNAs - Others (open for discussion) |
| ChIP-Seq | - Alignment Files in BAM format - List of Peaks in Excel format - List of Annotated Peaks in Excel format - BigWig File For Peaks Visualisation in Integrative Genomics Viewer (IGV) *Includes 1 pairwise comparison for every sample submitted for sequencing. Additional comparisons are welcome, please contact us for details. | - Others (open for discussion) |
| Human Exome Sequencing | - Alignment Files in BAM format - SNP / INDEL Files in standard VCF format - List of Annotated SNP / INDEL Files in MS Excel format *Please click here for the target file that we used for analysis. | - List of Annotated somatic SNP /INDEL in MS Excel format - Annotation against PHIAL in MS Excel format - Others (open for discussion) |
| Human Whole Genome Sequencing | - Alignment Files in BAM format - SNP / INDEL / CNV / SV Files in standard VCF format - List of Annotated SNP / INDEL / CNV / SV Files in MS Excel format *Analysis will be conducted using DRAGEN platform. *For somatic CNV calling, a matched normal sample is required. Please contact us for further details. | - Others (open for discussion) |
| Bisulfite Sequencing | - Alignment Files in BAM format (Human and Lambda if applicable) - List of Annotated CpG Methylation Sites in Text/Excel format - Bisulfite conversion rate of Lambda (if applicable) | - Others (open for discussion) |
| de novo Genome Sequencing | - de novo Assembly Files (raw files and contigs) in various formats - Predicted Coding Gene Files in Text format - List of Annotated Coding Gene in Text format - List of Repeats in Text format - List of non-coding RNA in Text format | - Circos Diagram for visualisation of the genome - Others (open for discussion) |
| de novo Transcriptome Sequencing | - de novo Assembly Files (raw files and contigs) in various formats - List of Transcripts in FASTA format - List of Annotated Transcripts (GO) in Excel format - Gene / Transcript Expression Level in Excel format - *List of Differentially Expressed Gene / Transcripts in Excel format *Includes 1 pairwise comparison for every sample submitted for sequencing. Additional comparisons are welcome, please contact us for details. | - Others (open for discussion) |
| Metagenomics Sequencing (Whole Genome Shotgun) | - de novo Assembly Files (raw files and contigs) in various formats - BLAST files (raw) in native BLAST format - Taxonomy Tree - Species Composition in Excel format | - Others (open for discussion) |
| Metagenomics Sequencing (16s Amplicon) | - Joined Paired-End Reads in FASTA format - OTU Table in BIOM format - *Taxonomy Summary - *Results of Alpha Diversity (diversity within sample group) - *Results of Beta Diversity (comparison of diversity between groups of sample) - *List of Differentially OTUs in Excel format *Includes 6 between group comparisons for every project. Additional comparisons are welcome, please contact us for details. | - Others (open for discussion) |
Pacbio Sequencing – Specific Deliverables Based On Project Type
For analysis needs beyond our routine analysis pipeline (standard deliverables), customized assistance could be provided (custom deliverables). Please note that the list below is by no means an exhaustive list of deliverables possible. Do contact us if you cannot locate what you need. Final deliverables are subjected to mutual agreement between CPOS and users.
| Type of Project | Standard Deliverables | Custom Deliverables |
| de novo Genome Sequencing | - de novo Assembly Files (raw files and contigs) in various formats - Predicted Coding Gene Files in Text format - List of Annotated Coding Gene in Text format - List of Repeats in Text format - List of non-coding RNA in Text format | - Circos Diagram for visualisation of the genome - Others (open for discussion) |
| Long-Range Structural Variation | - Alignment Files in BAM format - List of Structural Variations in Excel format | - Others (open for discussion) |
Downloads
Along with the analysis pipelines developed at CPOS, we have also accumulated resources that are useful for various types of data analysis. To facilitate other fellow bioinformaticians, these resources will be available through this website.
Exome Target Regions
The BED files were generated by merging the target and probe regions (provided by vendor), followed by adding 200bp padded region (100bp upstream and 100bp downstream). Conversion from hg19 to hg38 was done using Batch Coordinate Conversion (liftOver) from UCSC Genome Browser Utilities. The workflow is shown as below:
The padded combined regions in BED format can be downloaded by clicking the links below:

The padded combined regions in BED format can be downloaded by clicking the links below:
SeqCap EZ Exome + UTR1
[ hg19 MD5:2de06c08d21a97642c1a50ac609c32b2 ] [ hg38 MD5:c83c85ea28546ca58e06005ad752741d ]
1The original target and probe region files can be downloaded here.
xGen® Exome Research Panel v1.02
[ hg19 MD5:c79c2a7e3d9ba77b3c4f2f9e3eebce1b ] [ hg38 MD5:380d6ca15ee5d76d54ab1d843e31d49b ]
2The original target and probe region files can be downloaded here [MD5:1408942505230683c79955ddaae90e46].
Terms of Service
At the CPOS, we strive to apply the most suitable analysis methods to your dataset and that every analysis step is carefully planned before the start of the project. As CPOS provides free consultations which include advice on suitable analysis methods and tools, we urge users to discuss with us in advance. We will in good faith provide our advice based on our expertise, yet cannot be held fully liable for the most suitable analysis method. Agreement should be reached between CPOS analysis team and the project principle investigator with good mutual understanding to allow the sharing of responsibility.
Data Handling
All data is stored on networked storage drives with periodic back-ups but CPOS will not be responsible to store the data indefinitely. All data (raw data, analysis results and all intermediate files) housed by the CPOS Bioinformatics Core may be subject to deletion 1 month after data delivery to users without prior notice. Users are advised to keep a secure, clearly identified, permanent copy of the dataset.
Acknowledgement / Authorship
We appreciate your acknowledgement of the Centre for PanorOmic Sciences (CPOS) in your publications with data generated at our centre. Authorship is deem suitable if significant intellectual input is provided. Proper recognition documents the impact of our work and helps justify the continuation of subsidized services. Thank you very much.
Contact
bioinfo.cpos@hku.hk

Core Facilities
Address
6th Floor
The Hong Kong Jockey Club Building
for Interdisciplinary Research
5 Sassoon Road
Pokfulam, Hong Kong
Tel: 2831-5500
Fax: 2818-5653
Web: https://cpos.hku.hk
Email: enquiry.cpos@hku.hk
Office Hours
Mon-Fri: 9:00am – 5:30pm
Samples and goods reception not available 1:00pm – 2:00pm
Closed on Saturday, Sunday, all University and Public holidays.

